
Introduction to Genomics
Published:
A summary of genomic-related topics, containing notes I took from the Introduction to Genomics course from JHU, and Applied Computational Genomics course from Dr. Aaron Quinlan at U of Utah, with a splash of my understandings on these topics.
Table of Content
- DNA: the molecule holding our source code
- Structure of DNA
- Nucleotides building blocks (AGCT) stringed together.
- 3D structure: histones, chromatin and chromosome
- How is DNA sequenced
- Sequencing by synthesis/duplication of DNA
- DNA duplication: polymerase chain reaction (PCR)
- Next generation sequencing (NGS)
- Sequencing by “eavesdropping” each step of DNA synthesis
- Massive parallelization: billions of reads per run
- 150bp read length and pair end sequencing - Sequencing process - Sequencing error and base quality
- Central Dogma: DNA, RNA and proteins
- RNA, preRNA, mRNA
- Proteins
- Genome and genes
- Exon, intron, UTR, CDS, splicing, intergenic DNA
- Gene regulations/epigenetics
- Transcription factors, promoters, histones, CpG islands, DNA methylation
- Half of the genome are repeats
- Transposons, jumping genes and Barbara McClintock
- Stats & details of the human genome
- Ref genome build, SNP, INDEL, DNM, 98% chimps
- Stats summary
- NGS applications
- Whole genome/exosome seq, RNA-seq, ChIP-seq, methyl-seq, GWAS
- A bit of cell biology
- Eukaryotes, cell division, meiosis and recombination
- Indices and abbreviations
DNA: the molecule holding our source code.
Our body’s made of cells. It’s estimated that 40 - 150 trillion cells forms the body (excluding bactria in our gut). Different types of cells (e.g. muscle/liver/brain cells) form different organs/ parts of body, which carry out different functionalities - the heart pumps out blood into the circulation system, the stomach process food, the brain sends all sorts of magical top down control signals… These various departments assembled into the intricate machine that is our body.
Yet all these trillions of cells originated from a single cell - a fertilized egg. This cell is programmed to duplicate to produce more cells, and the cells are programmed to further differentiate to all types of cells.
There are many important questions to how the machine of the body was built and how each department carries out its functions:
- What are genes doing - what protein does a gene code? What’s the protein’s function?
- How are genes regulated by environments? Only 2% of genome codes for proteins, 95% of the genome is dedicated for regulation - when to turn a gene on or off (epigenetics).
- The regulation/ epigenetics might be the more important topic. Since it’s the directions / user manual that codes for how each cell react to environmental changes. Different types of cells (brain/skin/kidney cells, etc.) have the same genome, it’s the set of genes that are turned on/off that decides the function and the type of cell.
- When the cells malfunction and diseases occur, is caused by a bug in the code (gene alteration)? Or is it because the cells started to read the wrong set of directions (epigenetic, regulation of genes)?
- How are the genetic and epigenetic factors associated some of our traits, such as height, eye color, predisposition to diseases like Alzheimer?
By sequencing our DNA, we are hoping to answer questions like these by reading the source code and figuring out how our program works.
Structure of DNA
Deoxyribonucleic acid (DNA): nucleotides building blocks (AGCT) stringed together.
- 4 types of nucleotides building blocks AGCT: Adenine, Guanine, Cytosine and Thymine.
- A and G are purines (bigger), T and C are pyrimidines.
- DNA double-helix: 2 strings of poly nucleotide chains.
- The two chains are complimentary to each other, where an A always pairs with a T (and vice versa) in the other string, and a C paris with a G (and vice versa).
- The two chains form a twisted double-helix shape (like a revolving ladder), where each step is an A-T or C-G pair.
- A-T pairs are connected via 2 hydrogen bonds, where as C-G forms 3 hydrogen bonds.
- 5’ and 3’ end: each chain has a direction
- One end of the chain has an unreacted phosphate group at carbon 5 of the nucleotide, called 5’end.
- The other end has an unreacted -OH group at carbon 3 of the nucleotide, called 3’ end.
- DNA sequence is written from 5’ to 3’- a positive or + strand. Its reverse compliment generate the sequence of the paired chain - the negative strand (also from 5’ to 3’).
3D structure: histones, chromatin and chromosome
- Chromosomes contain all the DNAs in the cell nucleus. Fully stretched DNA is about 2 meters long. The double helix structure has to be tightly packaged in the nucleus.
- The double helix wraps around histones and form a “beads on string” structure, The histones pack together into chromatin fiber, that eventually lead to chromosomes.
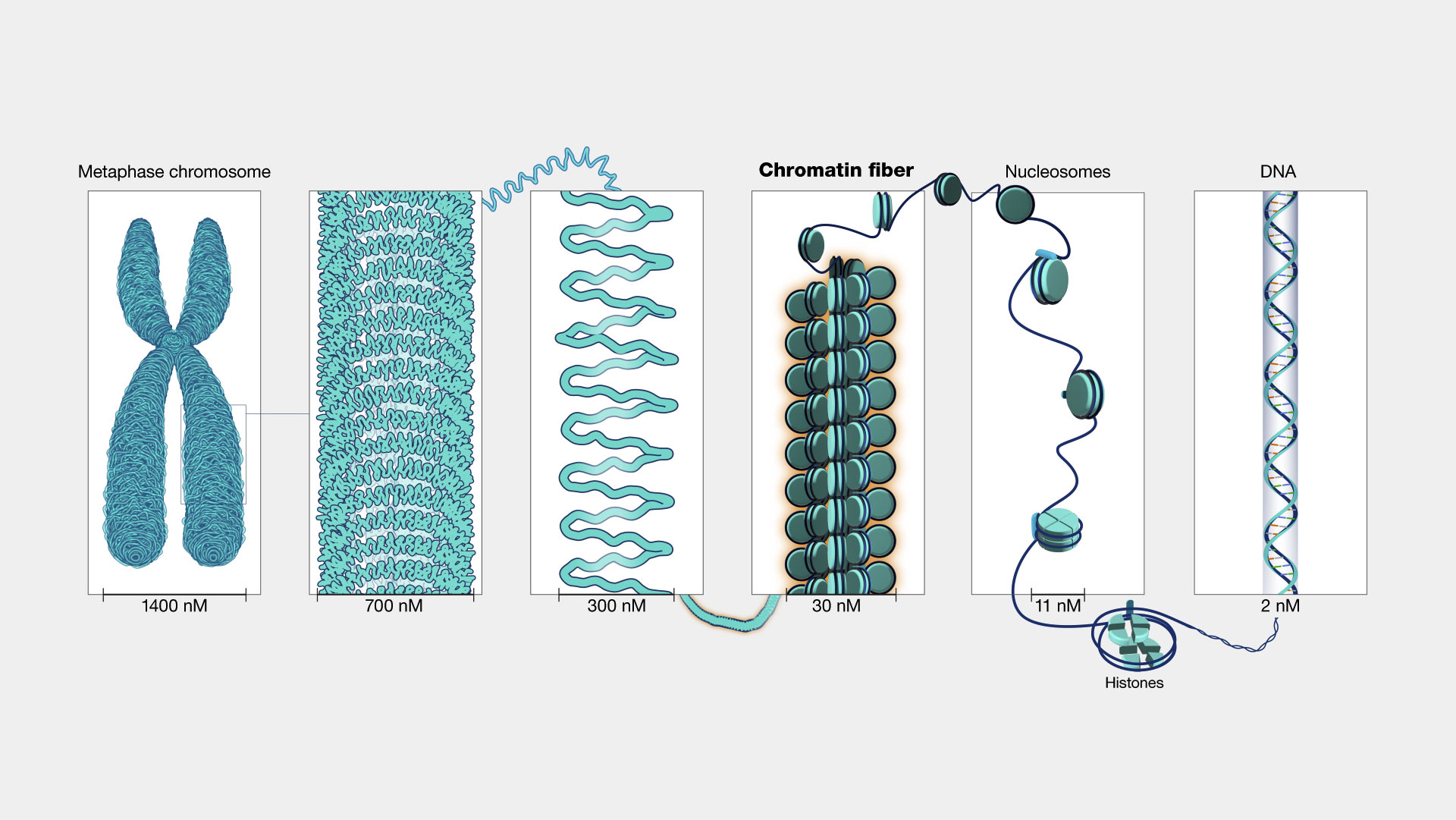
Source: https://www.genome.gov/genetics-glossary/Chromatin
How is DNA sequenced?
Sequencing by synthesis/duplication of DNA.
DNA can be duplicated very effectively. When a DNA is synthesized, if we could “eavesdrop” on the order of nucleotides are being added - we would learn the sequence.
This is achieved by attaching one nucleotide at a time during synthesis, and pause the reaction to read what nucleotide was just added, so when the entire DNA is synthesized, its whole sequence is read in the process.
DNA duplication: polymerase chain reaction (PCR)
- PCR technique allows rapid replications of DNA as well as laying the foundation of sequencing - sequence is read as DNAs are created.
- One pot chain reaction
- Reagents: DNAs to be replicated, primers, ATCG monomers, polymerase (proteins that stitch nucleotides together)
- Primer: short DNAs with 15 20 bases long that pair with ends of the DNAs to be duplicated. For example, the red lines at the top and bottom are two primers. It’s the reverse complement of the 3’ end (itself starts from 5’).
5' - CTATTCATT 3' - GATAAGTAAGTATGTGGTTCTAT......CTATCTATATCATATCTAACCCAGAC - 5' 5' - CTATTCATTCATACACCAAGATA......GATAGATATAGTATAGATTGGGTCTG - 3' ACCCAGAC - 5'
- Reaction cycle: 3 stages to duplicate each DNA chain in the system.
- Heating to 94 °C: DNA melts and two helix strands separate
- Annealing to 54 °C: the primers bind with full DNA chains (its harder for 2 full chains to find each other)
- Heating to 72 °C: polymerase fill in the missing part for each primer, the cycle ends.
- Cycle repeats.
- At the end of each cycle, number of full DNA chains is doubled. The system can go back to the first step by heating up again. After 30 cycles, it would produce 2^30 (about 2 billion) from the original one.
(A piece of irrelevant history: apparently the guy who invented PCR - Kary Mullis was tripping on LSD when he came up with the idea.)
Next generation sequencing (NGS)
The key is during DNA synthesize, only add one nucleotide at a time, pause the reaction, record that base, then add & record another.
The strength of NGS is massive parallelization - the ability to sequence billions of DNA at a time.
Sequencing by “eavesdropping” each step of DNA synthesis
- Imagine a DNA is a long Lego pillar like the picture below, where each small cubic building block is a nucleotide. 4 colors represent 4 types of bases AGCT.

Image adapted from Coursera Course: Algorithms for DNA Sequencing
- Sequencing starts by attaching DNAs to be sequenced (called templates) to a surface. There are 3 DNA templates / Lego pillars in the toy system (shown at top left corner of the picture), while in reality billions of DNAs would be sequenced at a time.
- Then specially modified nucleotides AGCT are added as reaction materials to synthesize/duplicate DNAs. The nucleotides are special in two ways: they have a terminator “cap”; and they fluoresce.
- The cap prevents another nucleotides (AGCT Lego cubes) from being added on to the capped nucleotide. This allows the DNA molecule to grow one nucleotide/base at a time, to pause the reaction so the current base can be read. The cap can be removed after the signal is taken.
- For example, at cycle 1, the three DNAs pillars have a paired strand of new DNA being synthesized right next to them, because of the capped AGCT cubes, only one cube was added to the new strand (paired according to A-T, C-G rule).
- These newly added nucleotide cubes fluoresce - more importantly, different types of nucleotides fluoresce different colors, which allows us to read the base identity by their color.
- A camera at top takes the fluorescent snapshot, and we learned what base is added. For example, the snapshot of cycle 1 tells us a G (red) is added to the top DNA being synthesized. We can infer the DNA being sequenced (the paired strand) has a C at the same position.
- Once the snapshot is taken, the nucleotides are washed out, the caps are removed, opening up the Lego cube for new cubes to be added in the next cycle. (In the picture showing the caps being removed, the original DNA pillars are not shown, but they are still attached to the surface).
- The cycle repeats, one capped nucleotide is added in the next cycle and a snapshot taken, the identity of the second nucleotides are read.
Massive parallelization: billions of reads per run
- Billions of DNA can be sequenced at the same time in a parallel fashion, by attaching billions of the Lego pillars and recording their signals all at once.
- In reality, the size of one DNA molecule is too small (~nm size) for the fluorescent light to be detected. Each DNA is duplicated to a cluster of itself to generate enough signals for a sequencer/human to read. An example of sequencing snapshot and base calling in each cycle is shown below.
 Image adapted from Applied Computational Genomics Course by Dr. Aaron Quinlan
Image adapted from Applied Computational Genomics Course by Dr. Aaron Quinlan
150bp read length and pair end sequencing
- NGS (Illumina) sequence DNA reads of ~150 base pair (bp) length (limited by errors such as fluorescence bleed through, more discussion later).
- 150bp is smaller than many repetitive segments of human genome, making them hard to place back (align) to the reference genome.
- One clever get around is prepare DNA samples of ~500 bp long, and sequence 75 bp on each end - pair end sequencing. An example is shown here, the first and last 75 bp of the sample are sequenced, with ~350 bp sequence in the middle missing.
5' - AACTTTAC(75bp)..../~350pb/....
..../~350pb/....TCCTTACA(75bp) - 3'
- The 75bp sequence from each end might align to thousands of loci (places) on the reference genome, but usually only one or two of them are within 500bp of each other.
Sequencing process
- All chromosomes from cell samples are shredded to ~500pb DNA strands (templates).
- Each DNA strand (template) is tethered to a slide, with billions of templates on the same slides.
- PCR duplicate each DNA template to a cluster of identical DNA strands, so that the fluorescence emitted by the cluster can be detected and read by sequencers.
- Add fluorescent labeled, terminator modified ATCG nucleotides. They will pair with the templates, and the pairing DNA strand extend only one base.
- Shine fluorescent light to read the current base.
- The terminator modification is reversed, and the pairing DNAs can be extended again, for the next nucleotide to be attached and read.
- Repeat until ideal length (150/75bp) of DNA is sequenced.
Sequencing error and base quality
- Out of sync bases
- Ideally the paired DNA adds one base at a time (on schedule), but some strands may miss a base (behind) or added more than one base (ahead). The out of sync bases can shine a different color and cause the signal to be less pure.
- The out of sync issue causes read quality to decrease in later cycles, and limiting read length to ~hundreds bp.
- Fluorescence bleed through between adjacent clusters
- Two clusters can get too close to each other, causing their fluorescence light/color to bleed over each other, making the color less pure and hard to identify.
- Quality score $Q=-10log(P_{err})$, where $P_{err}$ is the probability of the base identity is wrong.
- E.g. a base has 10% (P_err=0.1) chance of being a sequencing error, its quality score $Q=-10log(0.1)=10$
- Rule of thumb is Q>30 (P_err=0.001) - error of 1 in a thousand, is a high quality nucleotide.
Central dogma: DNA, RNA and proteins
- Central dogma states that information flows from DNA to RNA (transcription), and from RNA to proteins (translation).
- A gene usually refers to the segments of DNA that make proteins, which is ~2% of the genome.
- The central dogma not an absolute dogma. Information goes in the other direction, proteins coded by genes (transcription factors) can bind DNA and turn genes on/off.
- The on/off settings can determine a cells functions and types - whether a cell is a neuron or skin cell.
RNA, pre mRNA, mRNA
- Ribonucleic acid (RNA) is a single strand molecule made of a slightly different alphabet: Adenine, Guanine, Cytosine and Uracil (AGCU). Comparing to DNA, T is replaced by U.
- Another subtle (and often ignored) difference between RNA and DNA is as the name suggest: ribose vs dioxyribose sugar, there’s an extra -OH group attaching to the ribose ring at carbon 2 for RNA.
- RNA is transcribed from a gene region of DNA following the same pairing rules A-U, C-G.
- pre mRNA & mRNA: preRNA is the photocopy (1:1 mapping A-U, C-G) of the DNA sequence. preRNA goes through splicing processes, where chunks of sequences are deleted and/or rearranged (more on splicing later), resulting in matured/messenger RNA (mRNA).
Proteins
- Proteins are big molecules with various shapes. They conduct most of cell functions, including metabolism, digestion, delivery, signaling, structural support.
- Proteins are made of 20 types of amino acids as building blocks (comparing to 4 types of nucleotides for DNA and RNA), usually hundreds of amino acids long.
- Protein structure hierarchy: primary structure (AA sequence) -> secondary structure (alpha helix, beta sheet) -> tertiary structure (3d structure) -> quaternary structure (complex of protein molecules).
- Codon: When a matured RNA is translated to proteins, it is read 3 letters (a codon) at a time. Each codon maps to an amino acid identity.
- A codon is 3 nucleotides long, each nucleotide could any of the 4 choices (AGCU), resulting in 64 (4x4x4) combinations (codons). 61 codons correspond to amino acids, 3 are stop codons, indicating the end of a protein.
Genome and genes
- Genome
- Usually refers to the entire DNA string in nucleus.
- It’s ~3 billion base pair long, contains sequence of a single strand DNA (positive strand, goes from 5’ to 3’) from 23 chromosomes.
- Mitochondria has its own DNA (inherited from Mom); with length of ~1% of nucleus DNA.
- Blood cells don’t have a nucleus. Many liver cells are polypoid (more than 2 pairs of chromosomes).
- Genes: A very small portion (~2%) of the 3 billion bp genome encodes protein. These sequence segments are called genes or protein coding genes.
- Exons, introns, UTR, CDS:
- A gene contains a lot of sequence segments that don’t maps to amino acids (aren’t codons). The regions that codes protein are called exons, they are short segments separated by non-coding sequences called introns.
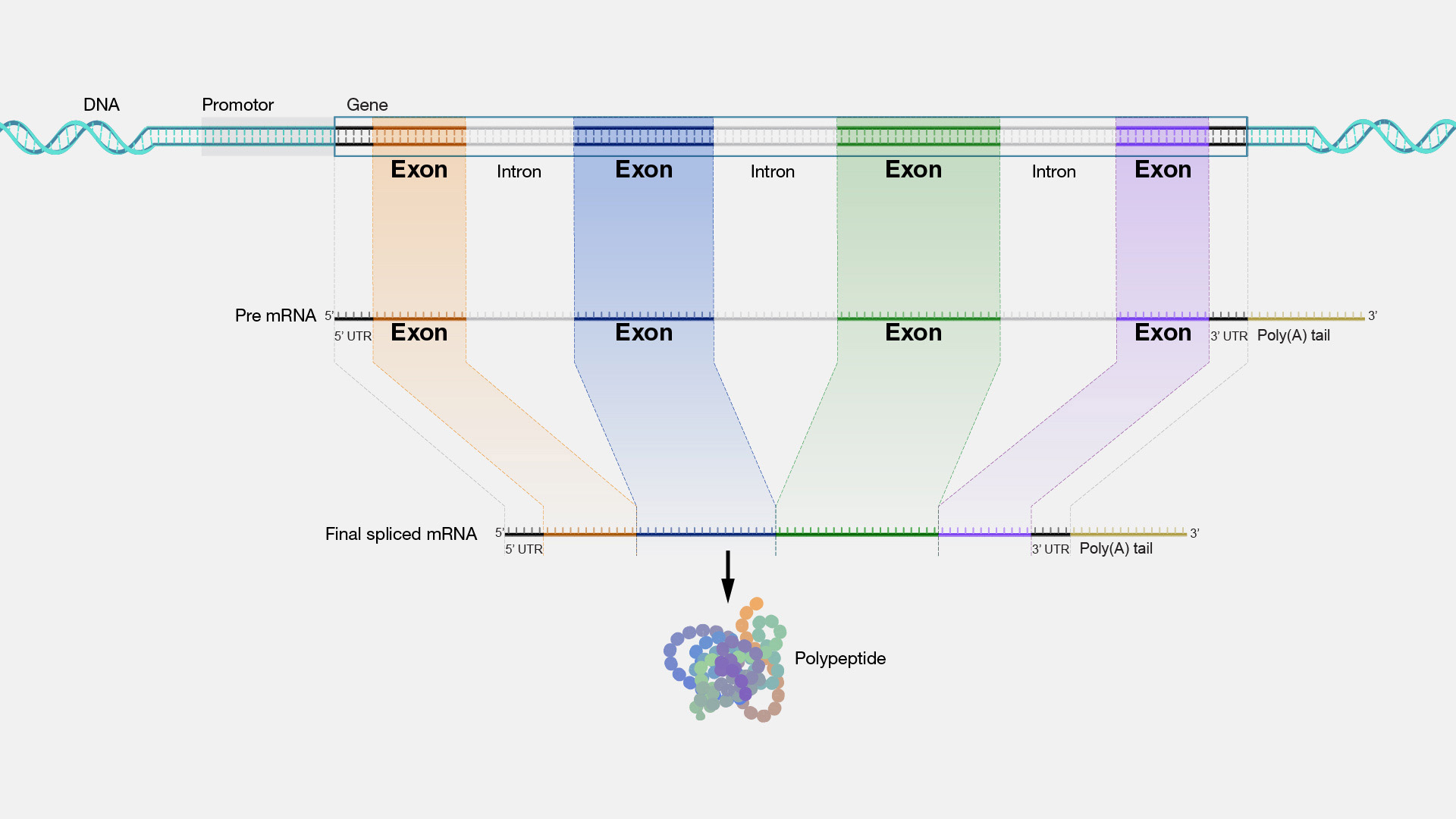 Image source: https://www.genome.gov/sites/default/files/media/images/2022-05/Exon.jpg
Image source: https://www.genome.gov/sites/default/files/media/images/2022-05/Exon.jpg - A gene is transcribed (via 1:1 mapping) to a pre mRNA. The pre mRNA contains all the non-coding sequences.
- Splicing: The introns then get cut out of the pre mRNA, and only exons remains in the matured / messenger RNA (mRNA).
- mRNA starts with untranslated regions (UTR) at 5’ end -> exons/coding sequences (CDS) -> 3’ UTR, then ends with a poly A tail.
- Alternative splicing: the exons can be shuffled around and ends up in a different order than they are in the original DNA sequences. This allows one gene to code multiple proteins.
- Over 90% of human genes undergo alternative splicing, meaning knowing the coding region does not directly lead to protein sequence.
- A gene contains a lot of sequence segments that don’t maps to amino acids (aren’t codons). The regions that codes protein are called exons, they are short segments separated by non-coding sequences called introns.
- Intergenic DNA:
- The stretches of DNA sequences that exist between genes on a chromosome.
- These sequences (comprise of 95% of the genome) do not code for protein or RNA, and were once thought to be “junk DNA” (yeah, like evolution would allow such huge waste of energy to survive the cruel selection process).
- These non coding regions are dedicated to regulate genes - when to turn a gene on or off, like the enhancers or silencers discussed in the following session.
Gene regulations/epigenetics
- Transcription factors & promoters
- Transcription factors are proteins that regulates the the transcription of genes. In a sense, they are the switches that turn a gene on or off.
- Promoters: for any gene to be expressed, the RNA polymerase needs to bind to a DNA region (upstream of the gene) called promoters to be able to start the transcription. In eukaryotes, the RNA polymerase can only bind to promoters with the help of transcription factors.
- Activators & enhancers: sometimes when transcription factors binds to DNA, it increase the likelihood of the gene being expressed (turn a gene on, or upregulate a gene). These transcription factors are called activators, and the DNA region they bind to are called enhancers.
- Repressors and silencers: conversely, when a transcription factor decreases the likelihood of a gene’s expression (turn it off, downregulate a gene), the transcription factor is called a repressor, and its binding DNA binding site is called a silencer.
- Histones
- Histones are the protein “beads” that DNA molecules wrap around in the “beads on string” structure in fig 1.
- When the histone beads pack tightly in chromatin, the transcription factors can’t access the promoter, and genes aren’t activated.
- The packaging has too loosen, so the promoter regions are exposed for gene expressions to occur.
- CpG islands
- CpG islands are regions of DNA in the genome that are rich in the dinucleotide CG (cytosine-phosphate-guanine, p represents the phosphate bond, means the direction is 5’ -> 3’). Note that the CG pairing rule means that for each CpG dinucleotide, there is a corresponding CpG dinucleotide in the reverse compliment string.
- CpG islands are usually located in or near the promoter regions of genes. CpG islands are found upstream of ~60% of protein-coding genes. In the early days of annotating the human genome, CpG islands were used as signposts of potential genes downstream.
- GC content (percentage of G or C in the string) is a predictor of gene content, and varies a lot in the genome. Fun fact, chromosome 19 has the highest GpC island density (~40/Mb), and has the highest gene density (~20/Mb) in the human genome.
- DNA methylation silencing genes
- For the CpG dinucleotides in CpG islands, the C (cytosine) in CG can be methylated.
- When many CpG were methylated in a CpG island, it’ll block the transcription factors’ binding, and inhibit or silence the expression of downstream genes.
- DNA methylation can be pretty stable. It can be passed on to daughter cells, but not inherited when a new baby is born.
- DNA methylation has been used as a biomarker for aging, and was associated with diseases such as cancer and developmental disorders.
Half of the genome are repeats
- Transposons are segments of DNA that can “jump” or “transpose” themselves to another position in the genome.
- They move by either insert copies themselves to a new location - the “copy/paste” mechanism for retrotransposons, or yeet themselves to another location in the genome - the “cut/paste” mechanism for DNA transposons.
- As a result of transposons copy pasting themselves allover the place, the human genome is littered with transposon repeats. It’s estimated that nearly half (45%) of the genome is comprised of transposable elements. The top 2 aggressive retrotranspons are L1 (comprises ~17% of genome) and Alu (~10%).
 Image source: left The impact of retrotransposons on human genome evolution | Nature Reviews Genetics, right Applied Computational Genomics Course by Dr. Aaron Quinlan
Image source: left The impact of retrotransposons on human genome evolution | Nature Reviews Genetics, right Applied Computational Genomics Course by Dr. Aaron Quinlan - Transposable genes were first discovered by the geneticist Barbara McClintock in maize in the 1950s, a discovery that completely destroyed her career at the time.
- Barbara McClintock: totally heroic figure, elected member of the National Academy of Science (highly unusual for a 40-year-old woman in 1940s). Her proposal in the 50s that “genes jump around” was met with great mockery and backlash. She stood her ground and continued research on her own for decades afterward, disappearing from the academia world. Until molecular techniques in 1980s caught up and showed her right. I’ll let the great Robert Sapolsky himself tell this palpitating tale.
The transposable elements in plants genome are a defense mechanism to the environmental challenges that induces cellular stress, because they can’t run away like animals, they developed fancier genetic tricks - juggling DNA segments (shuffling the deck) in hope of the recombination would generate something useful.
- Tandem & interspersed: repeating patterns
- Tandem repeats refer to (near) identical repeats occurring in a row (e.g. CAGCAGCAGCAGCAG…). The centromere comprise largely of ~180 base pairs long repeats that occur hundreds of thousands time in a row.
- Interspersed repeats refer to are located in different places in genome, sometimes across different chromosomes. The transposons are interspersed repeats.
Haploid, Diploid, Homo/hetero-zygous and some jargons
- Haploid and diploid
- Haploid: a single set of chromosomes in an organism’s cells.
- Diploid: humans (and other sexually reproducing organisms) are diploid. Each cell have 2 sets of chromosomes, one from each parent. The exception being the egg and sperm cells, they are haploid.
- Haplotype, genotype, homozygous and heterozygous
- At each site of the genome, we have a paternal and a maternal “haplotype”. The two alleles form our “genotype”.
- A homozygous site means the two alleles (haplotypes) are the same type, a heterozygous site means they are different.
- Karyotype
- A karyotype is a visual representation (usually by staining chromosomes and then pair them) of the complete set of chromosomes within a cell.
- Karyotyping is used to determine the number of chromosomes, to examine whether there are missing or extra chromosomes.
Stats & details of the human genome
- The first draft (“build 1”) of reference genome was released in 2001. We are currently on build 38.
- Last year (2022), the first complete, gapless, Telomere-to-Telomere (T2T) Consortium genome (3.055 billion bp) was released. The build 1 draft genome had many gaps (~80Mbp missing, with ~600 medically relevant genes in missing regions). After 2 decades of relentless work of filling the gaps, the genome was finally gapless (except for Y chromosome).
- Most of reference genome represents northern European male. There have been efforts to build more references with other ethnic origins.
- Comparing the genomes from any two humans, their genomes differ on average about one in every 700 bp (~0.14%), which leads to ~ 4 million bp differences for any two individuals.
- Single-nucleotide polymorphisms (SNPs) and Small Insertions/Deletions (INDELs)
- When comparing two genomes (strings), the differences can occur in 3 ways: substitution, insertion and deletion of one or more characters.
- Single-nucleotide polymorphism (SNP) refers to the difference where only one character (one nucleotide letter) is substituted by another in the genome.
- Small insertions/deletions (INDELs) refers to the difference where one or more characters are inserted or deleted in the genome.
- INDELs happen less frequently than SNPs and are harder to call.
- De Novo mutations (DNM)
- a mutation that occurs when sperms/eggs were produced. These are new mutations seen in the new generation but not in parents.
- DNMs occur ~30-40 per haploid genome (in one sperm or egg), meaning each offspring carries ~70 to 80 DNMs. Calculation: human mutation rate 1.1 x 10^-8 bp per generation, times 3.1 billion bp, leads to ~34bp per gamete cell.
- Total number of genes (still under debate): one estimate is ~20,000 protein coding genes (10% more than a worm or fly), and ~200,000 coding transcripts (isoforms of a gene that encodes a distinct protein).
- Human share 98% of DNA with chimpanzees. A more clarified statement is that when comparing human genes and chimp genes, 98% of them code for similar things - hormones, neurotransmitters, immune systems, and neither human nor chimp has genes for antlers, trunks, wings, fins, etc., similar claims are humans are 90% cats or 60% bananas.
- Humans have 2 sets of 23 chromosomes (23 pairs). 22 pairs of autosomes and 1 pair of sex chromosomes.
- The 22 autosomes are numbered according to their length: chromosome 1 is the longest (~250 Mb), and chromosome 21 is the shortest. Chromosome 22 was thought to be shorter than 21 at the time, when the length was determined by staining the chromosomes and look at them under microscopy, which is quite imprecise and chromosome 21 was actually slightly smaller than 22.
Stats summary
| Human Genome | Stats |
|---|---|
| Whole Genome | 3.055 billion bp |
| All Exons | 60 Mb (2%) |
| Internal (1) exon | hundreds bp (ave. 145b) |
| Intron | thousands bp (ave. 3.3Kb) |
| Coding Seq (CDS) | ave. 1.3 kb |
| 5’ UTR | ave. 300 b |
| 3’ UTR | ave. 770 b |
| 1 gene | ave. 27 kb |
| Genes | 20,000 |
| Transcripts | 200,000 |
| SNP/INDEL | 4 million (1/700b) |
| De novo mutation | ~35/gamete |
| GC content | 35-60% for 100Kb, ave. 41% |
| Transposable elements | 45% of genome |
| Mitochondria | 30 Mb (1%) |
NGS applications
- Whole genome sequencing (WGS)
- Sequencing the entire genome (~3.1 billion bp) - what took the Human Genome Project 10 years and 3 billion dollars to achieve, now can be done overnight for a thousand bucks.
- Whole exosome sequencing (WES)
- Sequencing all exons - all protein coding genes. Since exons are only ~2% of the genome (~60 million bp), it’s a lot cheaper and faster.
- RNA sequencing (RNA-seq)
- To find out which genes are turned on for cells. (as not all exons, or genes are turned on for a certain cell)
- Transcribed mRNA has a string of AAAAAs attached to it. The polyA tail can be captured by a string of Ts.
- The captured mature RNA can be reverse transcribed into complementary DNA (cDNA) by reverse transcriptase. The DNA is then sequenced.
- ChIP-seq
- To find the DNA regions where transcription factor proteins bind to, which gives information of which genes are turned on/off.
- First let proteins bind to DNA. Fragment the DNA, then pull the proteins with antibody pulldown, with DNA fragments stuck to the proteins. Remove the protein, then sequence DNA.
- Bisulfate sequencing (methyl-seq)
- To find places of DNA methylation.
- The methyl -CH3 group attaches to C in CpG islands. The methylation can be inherited by next generation daughter cells.
- Take two DNA samples. Treat one sample with bisulfate conversion, which turns all the Cs to Us, except the Cs that are methylated.
- Sequence both sample and compare. The converted sample needs a special aligner since most Cs are turned to Us, and it won’t align well with the normal reference.
- Genome-wide association studies (GWAS)
- GWAS are not a sequencing technique. It’s an important application of NGS.
- It searches for association between genetic variants (genotypes) and traits (phenotypes) by comparing genomes in large populations.
- Diseases are a common category for GWAS studies. GWAS can provide insights into the genetic basis of diseases and information about drug targets and therapeutic/treatment strategies.
A bit of cell biology
- Eukaryotes, archaea and bacteria are the 3 domain system of organism classification. Archaea and Bacteria are sometimes group together and called prokaryotes, because they both lack nucleus and other membrane bound organelles (such as mitochondria).
- Eukaryotes: organisms whose cells have a nucleus, which sequesters DNA inside.
Archaea and bacteria (prokaryotes) don’t have a nucleus, no chromosomes. DNA is loosely organized.
- Evolutionarily, eukaryotes and prokaryotes split near the beginning of the evolution of life on earth, bacteria and archaea split later.
Mitochondria is considered to be absorbed by the ancestor of all eukaryotic cells, becoming part of every eukaryote.
- The cell (division) cycle: the series of events that take place in a cell that cause it to divide to 2 daughter cells.
| State | Phase | Abbreviation | Description |
|---|---|---|---|
| Resting | Gap 0 | G_0 | Cells not dividing |
| Interphase | Gap 1 | G_1 | Cell growth. G1 checkpoint ensures cell is ready for DNA synthesis |
| ^ | Synthesis | S | DNA replication |
| ^ | Gap 2 | G_2 | Growth & prepare for mitosis. G2 checkpoint ensures cell is ready for mitosis phase and divide. |
| Cell division | Mitosis | M | Chromosome separation. Consists of prophase, prometaphase, metaphase, anaphase and telophase. Metaphase checkpoint ensures cell is ready to complete division. |
- Stem cells can differentiate into different types of cells, which involves another type of division where the 2 daughter cells are not identical to each other.
- Meiosis (reduction division): a special type of division in reproduction process that produces gametes, such as sperm or egg cells.
- Recombination/crossing over: prior to the meiosis division, genetic material from the paternal and maternal copies of each chromosome is crossed over, creating new combinations of code on each chromosome. It is estimated to happen one crossover read per chromosome. It causes our chromosomes to be not identical to either of our parents’ chromosomes. The randomness introduced by recombination is one of the reasons that children of the same parents are not identical.
Indices and abbreviations
- 3’ end
- 5’ end
- CDS (coding sequence)
- ChIP-seq
- CpG islands
- DNM (De Novo mutation)
- DNA double-helix
- DNA methylation
- DNA nucleotides AGCT
- DNA polymerase
- GC content
- GWAS (genome wide association study)
- INDEL (small insertion/deletion)
- Methyl-seq (bisulfate sequencing)
- NGS (next generation sequencing)
- NGS DNA cluster
- NGS DNA template
- NGS terminator
- PCR (polymerase chain reaction)
- PCR primer
- RNA polymerase
- RNA splicing
- RNA-seq
- SNP (single nucleotide polymorphism)
- UTR (untranslated region)
- Whole exosome sequencing (WES)
- Whole genome sequencing (WGS)
- activator
- archaea
- autosome
- bacteria
- cDNA
- central dogma
- chromatin
- chromosome
- codon
- diploid
- enhancer
- epigenetics
- eukaryotes
- exon
- gene
- genome
- haploid
- heterozygous
- histones
- homozygous
- human mutation rate
- intergenic DNA
- interspersed repeat
- intron
- karyotype
- mRNA (messenger/mature RNA)
- meiosis (reduction division)
- mitochondria DNA
- negative strand
- pair end sequencing
- positive strand
- preRNA
- prokaryotes
- promoters
- protein
- quality score
- recombination/crossing over
- reference genome
- repressor
- sequencing error
- silencer
- stem cell
- stop codon
- tandem repeat
- transcription
- transcription factors
- translation
- transposable genes
- transposons