Exact matching algorithms like the Naive Exact Matching, Boyer-Moore Matching Algorithm and Index assisted matching can align a pattern P (sequence read) to a text T (genome).
However, in real life, the sequence read will rarely be an exact match to the genome. Two main reasons that cause a sequence read to deviate from the reference genome are 1) sequencing errors and 2) individual variations. A big part of computational genomics is to distinguish true biological variations from noises like sequencing errors.
Variations: substitution, insertion and deletion
There are 3 types of variations when comparing a sequence to a reference genome: - Substitution: one letter is substituted by another. - Insertion: there’s an extra letter in the sequence read, or a gap in reference genome. - Deletion: oppositely, there’s an extra letter in reference genome, or a gap in the sequence read.
T: GGAAAAAGAGGTAGCGGCGTTTAACAGTAG ||||||||| P: GTAACGGCG ↑ Mismatch/Substitution T: GGAAAAAGAGGTAGCG-GCGTTTAACAGTAG |||||| ||| P: GTAGCGGGCG ↑ Insertion T: GGAAAAAGAGGTAGCGGCGTTTAACAGTAG || ||||||| P: GT-GCGGCG ↑ Deletion
A substitution is also called a mismatch or mutation, and insertion and deletion sometimes are bundled and referred to as an INDEL.
Hamming and edit distance
To quantify how different two string X and Y are, we calculate the distance between the two strings. There are different types of string distances or metrics that can be used, depending on the context and purpose of the comparison.
Hamming distance: the minimal number of substitution needed to turn one string into the other.
Hamming distance only considers the cases of mutation, and requires X and Y to be of the same length.
X: GAGGTAGCGGCGTTTAAC | |||| ||| ||||||| Hamming distance=3 Y: GTGGTAACGGGGTTTAAC
Edit/Levenshtein distance: the minimal number of edits (including substitutions, insertions and deletions) needed to turn one string into the other.
When measuring edit distance, X and Y does not need to be the same length.
X: GAGGTAGCGG-GTTTAAC | |||| || ||||||| Edit distance=4 Y: GTGGTA--GGGGTTTAAC
Approximate matching with the pigeonhole principle
There’s a framework that can adapt any exact matching algorithm to solve an approximate matching problem.
First let’s think about a simple case, where up to 1 edit is allowed in pattern P for the matches to occur.
- The trick is to split P into two partitions u and v, and either u or v has to match exactly to T - the edit can only exist in one of them, hence the other has to be an exact match.
- We can then use the exact matching algorithm to find locations where either u or v is an exact match, and verify whether it leads to a true match within the 1 edit criteria.
Generally, to find matches where n edits are allowed, we can split P into n+1 partitions/ segments, search for hits where any partition is an exact match, and verify the other partitions.
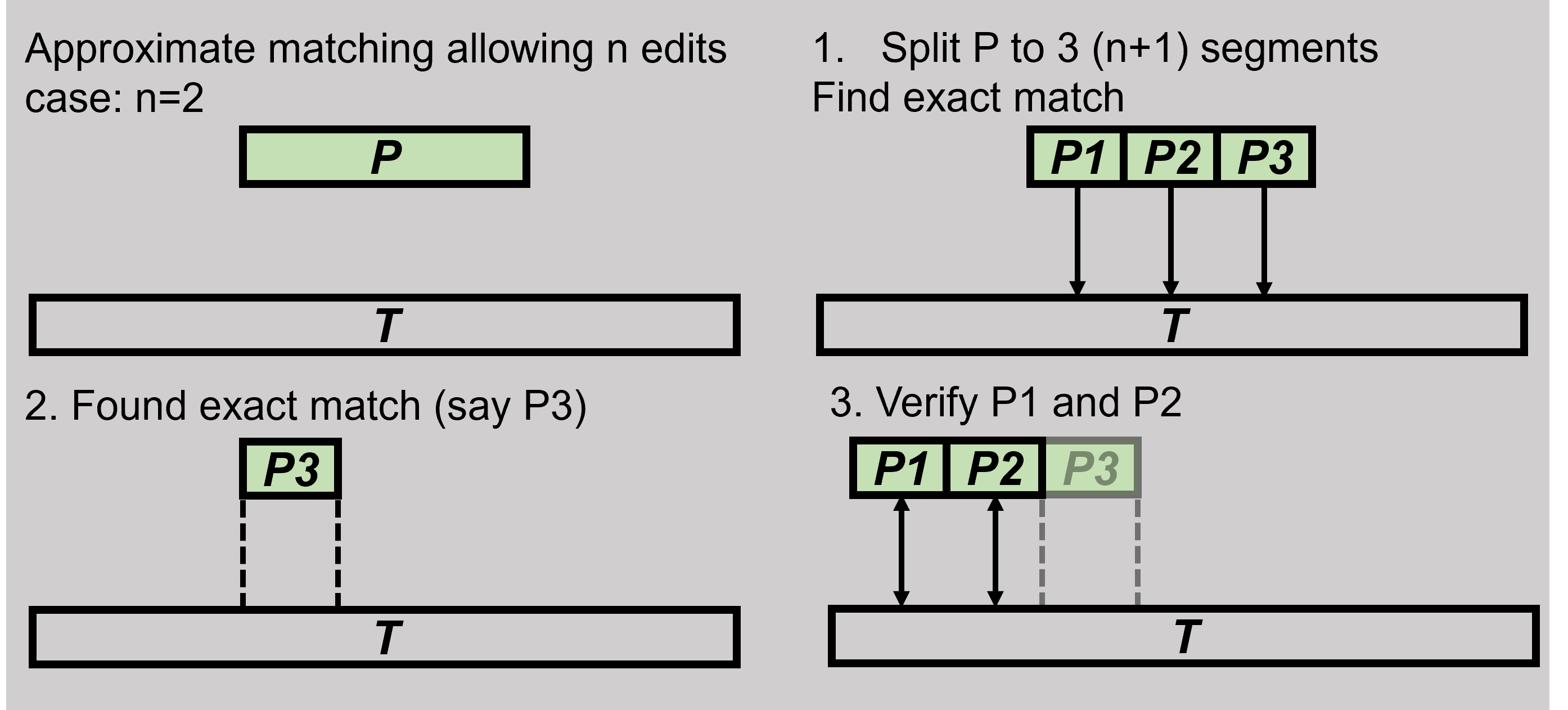
This is called a pigeonhole principle: if you place 10 pigeons in 9 holes, at least 1 hole will contain 2 pigeons.

Our case is slightly different - we are placing edits in the partitions, and we have more partitions (n+1) than edits allowed (n), at least one partition will be empty (without any edits) and have to be an exact match.
So in our case there are more holes than pigeons, and if we place 8 pigeons in 9 holes, at least 1 hole will be empty.
Code example - approximate matching with Boyer-Moore
Here’s an example of adapting the Boyer-Moore algorithm to achieve approximate matching, based on the pigeonhole principle. The code is in Python, the code for the Boyer-Moore class can be found at http://d28rh4a8wq0iu5.cloudfront.net/ads1/code/bm_preproc.py
def approximate_match_boyer_moore(p, t, n):
# Pigeonhole: divide p to n+1 segments
seg_len = int(round(len(p) / (n+1)))
all_matches = set() # avoid recording the same match from multiple segment at same alignment/offset position
for i in range(n+1): # loop trough each segment p[i * seg_len, (i+1) * seg_len], last segment edge case
start = i * seg_len
end = min( (i+1) * seg_len, len(p) ) # the last segment end should not surpass len(p)
seg = p[start:end]
seg_bm = BoyerMoore(seg)
seg_matches = boyer_moore(seg, seg_bm, t)
# check if rest of p matched within the allowed mistake counts
for m in seg_matches:
if m - start < 0 or m - start + len(p) > len(t):
continue # p goes out of t in this alignment
mismatches = 0
for j in range(start): # check p[:start]
if p[j] != t[m - start +j]:
mismatches += 1
if mismatches > n:
break
for j in range(end, len(p)): # check p[end:]
if p[j] != t[m - start +j]:
mismatches += 1
if mismatches >n:
break
if mismatches <= n:
all_matches.add(m - start)
return list(all_matches)